www.thetinman.org Copyright © All rights reserved.

Disclaimer: The material presented in this site is intended for public educational purposes only. The author is not offering medical or legal advice. Accuracy of information is attempted but not guaranteed. Before undertaking any diet, or health improvement program, you should consult your physician. The author is in no way liable or responsible for any bodily harm, physical, mental or emotional state of any patient reacting to any of the content on this site. Thetinman.org has not examined, reviewed or tested any product or service mentioned herein. We are not being paid to advertise or promote any product or service mentioned herein. The links are offered strictly as examples of resources available. The site assumes no responsibility or liability of any kind related to the content of external sites or the usage of any product or service referenced. Links to external sites were live at the time of creation of the link. Thetinman.org does not create content for or manage external sites. The information can be changed or removed by the external site’s administrators at any time and they are responsible for the veracity of their information. Links are provided to support our data and supply additional resources. Please report broken links to administrator@thetinman.org. Thetinman.org is not a charitable foundation. It neither accepts nor distributes donations or funds of any kind.


Perhaps the most important update comes from a review from Dr. Marinos Dalakas, formerly of the National Institutes of Health, and one of the most off-cited experts on stiff-person syndrome.
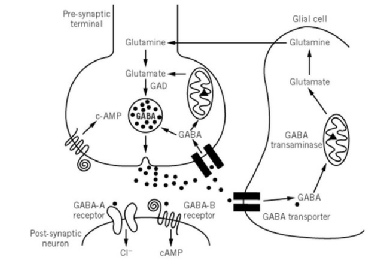
It was believed that the antigens that inhibit enzymes (GAD) that catalyze decarboxylation of glutamate to gamma-aminobutyric acid (GABA) were responsible for SPS.
GABA is the main inhibitory central nervous system neurotransmitter and regulates muscle tone. This was the reason for the initial focus on the antibodies as a causative factor and has been the focus of extensive research. The fact that stiff-person syndrome symptoms improve with GABA-enhancing drugs suggests that further research is needed to understand the correlation with the GABA system.
However Anti-GAD65 antibodies have not been proven to cause stiff-person syndrome or correlate to symptom severity or progression of the disease.
There is also correlation between stiff-person syndrome and autoantibodies to GABARAP, amphiphysin, gephyrin, and several MHC class-II alleles: Dqβ1 and DRβ1. No single predominant allele has been identified.
Anti-GAD65 antibodies are found in one to two percent of the general population and in five percent of patients with other neurological disorders.
They are found in patients with Graves’ disease, Hashimoto’s thyroiditis, myasthenia gravis, Lambert-Eaton syndrome, pernicious anemia, vitiligo, cerebellar ataxia, seizures, limbic encephalitis, brainstem encephalitis, opthalmoplegia, parkinsonism, myelopathy and are found in thymoma, Hodgkin’s lymphoma, renal-cell, breast, small-cell lung, and colon cancers.
The factor that causes GAD65 autoimmunity has not yet been identified. Anti-GAD65 antibodies are found in the serum and cerebrospinal fluid in 60 to 80% percent of reported stiff-person case studies. Anti-GAD67 antibodies are found in less than half and at much lower titers.
Up to one-third of stiff-person patients are diagnosed with autoimmune type 1 or type 1.5 (LADA) diabetes, which either precedes the onset of SPS or develops during the course of the disease. The number of patients with type 1 diabetes that go on to develop SPS is a fraction of a percentage. Autoimmune thyroid disease is also found in a large percentage of SPS patients. In the very beginning, Drs. Moersch and Woltman suggested that stiff-person syndrome had a metabolic basis. Connection with other autoimmune endocrine diseases suggests that there are certainly similar mechanisms at work.
GABA is expressed in the pancreas and anti-GAD65 antibodies are found in 70 - 80% of T1 and T1.5 diabetes patients. The GAD65 antibody test is used to distinguish Type 1 and 1.5 diabetes from Type 2. Type 1 diabetes patients show values of less than or equal to 20 IU/ml (ELISA) whereas SPS patients showed values of greater than or equal to 20 IU/ml.
Stiff-person patients show higher level IgG2 and lower IgG4 levels than T1D patients. They were found to have a higher titer of GAD65 and higher binding frequency to IgG1-4 b78 subclass frequencies than patients with T1D. No differences were found between SPS and T1D in the IgG1-4 subclass frequencies b96-11. The T1D GAD epitopes recognize conformational epitopes, whereas the GAD65 antibodies found in SPS recognize linear and denatured epitopes. This level of testing is only performed in a research setting. The important fact is that they are similar but not the same.
Both GAD65 and GAD67 are expressed in the thymus. The nervous and immune systems demonstrate reciprocal regulatory relationships via shared chemical messengers through the hypothalamic-pituitary-adrenal axis and for the autonomic nervous system via an anatomic connection where nerve terminals end in peripheral immune organs. There are receptors for neurotransmitters, neuropeptides, and hormones on immune cells. Neural cells have receptors for cytokines. It is not yet clear whether the signaling mechanisms used in the CNS are replicated in the immune system.
Muscles are also secretory organs that produce cytokines and other peptides that exert autocrine (chemical messengers that bond to receptors on the same cell), paracrine (cells that produce a signal to induce changes in nearby cells), and endocrine effects called myokines.
Myokines may influence cancer cell growth and pancreas function. Many proteins produced by skeletal muscle are dependent upon contraction; therefore, physical inactivity probably leads to an altered myokine response, which could provide a potential mechanism for the association between sedentary behavior and many chronic diseases.
Autocrine agents, which include cytokine, interleukin-1, and monoclonal T and B cells, are instrumental in cell-mediated cytoxicity and tumor formation.
Paracrine agents (including growth factors, clotting factors, responses to allergens,
tissue repair, and formation of scar tissue) regulate insulin, pituitary, gonadal,
and hypothalamic hormones.
Endocrine hormones affect muscle function. Insulin, glycogenesis and glycolysis occur in the muscles. Cortisol inhibits glucose uptake in the muscle. Adrenalin and noradrenaline boost oxygen to the muscles and increase skeletal muscle readiness.
Testosterone affects anabolic muscle growth, increasing muscle mass, strength and bone densiy. High cortisol over time can cause muscle atrophy. Estrogen accelerates metabolism but reduces muscle mass.
In some - but not all - cases, stiff-person patients responded to treatment with IVIG, plasmapheresis, and immunomodulation drugs. However, despite extensive research, causation due to a specific autoantigen has not been proven.
1. It is possible that stiff-person syndrome is a T-cell mediated disease. In a small study (eight patients), T-cells from GAD65 epitopes aa81-171 and aa313-403 were found. In type 1 diabetes, the epitopes found were aa161-233 and aa473-555. The autoantibodies were mostly IgG1 and IgG3, suggesting a Th1 helper T-cell response. T-cell response could drive the disease in the early stages. At autopsy, no T-cell infiltrations have been found in the CNS of stiff-person patients.
2. Autoantibodies to GAD65 (anti-GAD65 are present in the sera of 70–80% of patients with type 1 diabetes (T1D), but antibodies to the structurally similar 67 kDa isoform GAD67 are rare. Antibodies to GAD67 may represent a cross-reactive population of anti-GAD65 , but this has not been formally tested.
3. The paraneoplastic variant is associated with anti-amphiphysin and anti-gephyrin antibodies. These antibodies have no clear pathogenic role.
4. GABA receptor-associated protein (GABARAP) interacts with gephyrin to assemble the GABA-α receptors. GABARAP and GAD65 antibodies coexist in up to 70% of stiff-person patients. GABARAP down-regulates the density of GABA-α receptors in the neuronal processes of the hippocampal neurons. It is not yet clear whether it is causative. Unlike GAD antibodies, in a small case study (eight patients), the level of GABARAP antibodies correlated to symptom severity.
5. Another possible culprit could be dysfunction of the GABA receptors. An inflammatory process could lead to release of pre- and post-synaptic antigens. Stiff-person syndrome related antibodies seem to cause dysfunction rather than destruction of the synapses.
6. A very low concentration of IgG in the brain parenchyma can affect synaptic transmission. If the main antigen is expressed in the brain, it could constantly stimulate resident B-cells to produce antibodies. B-cells cross the blood brain barrier into the brain. The intrathecal antibody synthesis in the CSF recognizes different epitopes than those found in the serum. More study is needed to determine how the antibodies, after crossing the blood-brain barrier or synthesized intrathecally, could penetrate vesicles or neurons and block their function or the synthesis of GAD.
7. In limbic encephalitis, the autoantibodies attack extra-cellular NMDA and AMPA receptors. GAD65 peptide fragments could present on the neuronal surface and act as a target for antibodies.
8. GAD could also be attacked by the immune system due to membrane association with the vesicles through heat shock proteins.
9. Mutations of the GLRA1 glycine-receptor are the basis for startle disease and could correlate to the stiff limbs and startle aspect of stiff-person syndrome.
10. A recent study showed that mice possessing a monoclonal GAD65-specific CD4+ T cell population (4B5, PA19.9G11, or PA17.9G7) develop a lethal encephalomyelitis-like disease in the absence of any other T cells or B cells. GAD65-reactive CD4+ T cells were found throughout the CNS in direct concordance with GAD65 expression and activated microglia: proximal to the circumventricular organs at the interface between the brain parenchyma and the blood-brain barrier. In the presence of B cells, high titer anti-GAD65 autoantibodies were generated, but these had no effect on the incidence or severity of disease. In addition, GAD65-specific CD4+ T cells isolated from the brain were activated and produced IFN-g. These findings suggest that GAD65-reactive CD4+ T cells alone mediate a lethal encephalomyelitis-like disease that may serve as a useful model to study GAD65-mediated diseases of the CNS.
11. In one study, rituximab penetrated the intact blood brain barrier, but its concentration in the cerebral spinal fluid (CSF) was 600–1000-fold lower than in serum. Therefore, the depletion of CSF B cells may not be as efficient as for peripheral blood B cells and as a consequence, CSF may serve as reservoir for GAD65-specific IgG memory B cells. GAD65 antibodies were present both in serum and in CSF prior to rituximab treatment. There was no access to CSF samples after treatment, so the persistence of symptoms could also be explained with intramedullar or intrathecal plasma or memory cells, which were spared during anti-CD20 treatment and continued to produce GAD65 antibodies.
12. There have been two reported cases of congenital stiff-person syndrome. A genetic connection has yet to be clearly identified. A connection to HLA-B27 has been found in the presence of other autoimmune diseases such as ankylosing spondylitis, sero-negative rheumatoid arthritis, inflammatory bowel disease, and psoriatic arthritis.
*Source: Alexopoulos H, Dalakas MC. A critical update on the immunopathogenesis of stiff-person syndrome. Eur J Clin Invest. 2010;40(11):1018-1025. doi: 10.1111/j.1365-2362.2010.02340.x.
13. Is there a genetic connection?
Patients with SMS carried the IDDM-protective DQB1*0602 allele and other sequence-related DQB1*06 alleles with the same frequency observed in controls. In contrast, these alleles are rarely found in IDDM. Five of eight (62.5%) SMS patients lacking a DQB1*06 allele were diabetic in contrast to only 2 of 10 (20%) with a DQB1*06 allele (P = 0.08), suggesting that the presence of DQB1*0602 or other DQB1*06 alleles may be associated with a reduced prevalence of diabetes among patients with SMS.
Pugliese A, Solimena M, Awdeh ZL, et al. Association of HLA-DQB1*0201 with stiff-man synrome. J Clin Endocrinol Metab. 1993 Dec;77(6):1550-3. Link to article.
WHAT CAUSES STIFF-PERSON SYNDROME ?
Disclaimer: The material presented in this site is intended for public educational purposes only. The author is not offering medical or legal advice. Accuracy of information is attempted but not guaranteed. Before undertaking any diet, or health improvement program, you should consult your physician. The author is in no way liable or responsible for any bodily harm, physical, mental or emotional state of any patient reacting to any of the content this site. Thetinman.org has not examined, reviewed or tested any product or service mentioned herein. We are not being paid to advertise or promote any product or service mentioned herein. The links are offered strictly as examples of resources available. The site assumes no responsibility or liability of any kind related to the content of external sites or the usage of any product or service referenced. Links to external sites were live at the time of creation of the link. Thetinman.org does not create content for or manage external sites. The information can be changed or removed by the external site’s administrators at any time and they are responsible for the veracity of their information. Links are provided to support our data and supply additional resources. Please report broken links to administrator@thetinman.org.